Cusabio Tag Recombinants
Tag antibodies Tag antibodies can be used for the detection and purification of multiple proteins with the same tag. When
Read More
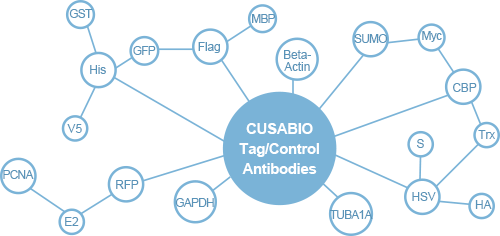
Tag antibodies Tag antibodies can be used for the detection and purification of multiple proteins with the same tag. When
Read More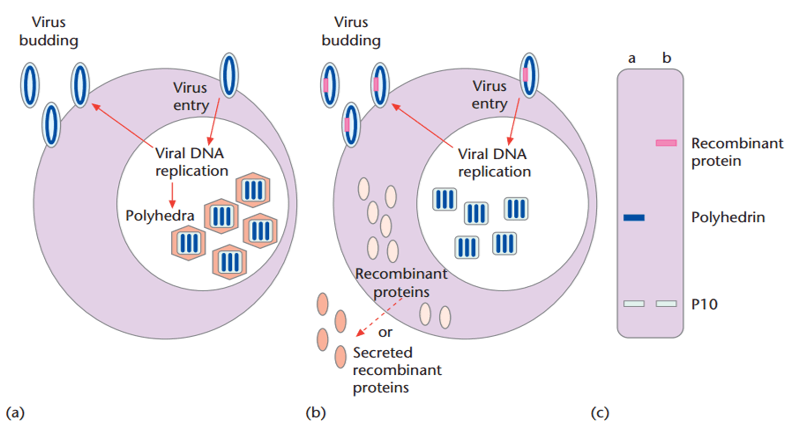
Abstract Baculoviruses are lethal insect pathogens, predominantly of the order Lepidoptera. These viruses have a biphasic life cycle, which greatly
Read More
Purity: greater than 95% as determined by SDS-PAGE. Endotoxin: Less than 1.0 EU/μg as determined by the LAL method. Activity
Read More
Description For rapid detection of IgG and IgM anti-SARS-CoV-2 (2019-nCoV) in 10-15 minutes during Covid-19. COVID-19 (coronavirus disease) is an
Read More
General description Direct detection of SARS-CoV-2 viral proteins (antigens) in nasal swabs and other respiratory secretions using lateral flow immunoassays
Read More
Description MAXgene GMP Transfection Reagent is a cGMP transfection reagent for the development and manufacture of viral vectors for cell
Read More

The global outbreak and pandemic of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have a significant impact on
Read More
Seahorses have a circum-global distribution in tropical to temperate coastal waters. Yet, seahorses present many adaptations for a sedentary, cryptic
Read More
The vertical distribution of subseafloor archaeal communities is regarded as primarily managed by in situ situations in sediments similar to
Read More