
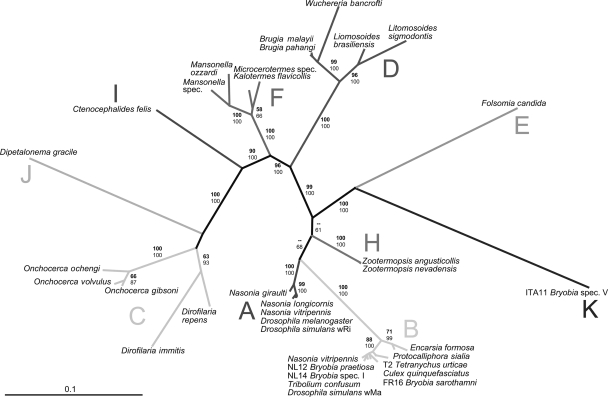
How diverse is the genus Wolbachia? Multiple-gene sequencing reveals a putatively new Wolbachia supergroup recovered from spider mites (Acari: Tetranychidae).
At least 20% of all arthropods and a few nematode species are contaminated with intracellular micro organism of the genus Wolbachia. This extremely diverse genus has been subdivided into eight “supergroups” (A to H) on the foundation of nucleotide sequence information.
Here, we report the discovery of a new Wolbachia supergroup recovered from the spider mite species Bryobia species V (Acari: Tetranychidae), primarily based on the sequences of three protein-coding genes (ftsZ, gltA, and groEL) and the 16S rRNA gene.
Other tetranychid mites possess supergroup B Wolbachia strains. The discovery of one other Wolbachia supergroup expands the identified range of Wolbachia and emphasizes the excessive variability of the genus.
Our information additionally make clear the present supergroup construction and spotlight the use of a number of gene sequences for sturdy phylogenetic evaluation. In addition to earlier experiences of recombination between the arthropod-infecting supergroups A and B, we offer proof for recombination between the nematode-infecting supergroups C and D.
Robust delineation of supergroups is important for understanding the origin and unfold of this widespread reproductive parasite and for unraveling mechanisms of host adaptation and manipulation throughout a big selection of hosts.
Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast most cancers.
Inherited predisposition to breast most cancers is identified to be brought on by loss-of-function mutations in BRCA1, BRCA2, PALB2, CHEK2, and different genes concerned in DNA restore. However, most households severely affected by breast most cancers don’t harbor mutations in any of those genes.
In Finland, founder mutations have been noticed in every of those genes, suggesting that the Finnish inhabitants could also be a wonderful useful resource for the identification of different such genes.
To this finish, we carried out exome sequencing of constitutional genomic DNA from 24 breast most cancers sufferers from 11 Finnish breast most cancers households. From all uncommon damaging variants, 22 variants in 21 DNA restore genes had been genotyped in 3,166 breast most cancers sufferers, 569 ovarian most cancers sufferers, and a couple of,090 controls, all from the Helsinki or Tampere areas of Finland. In Fanconi anemia complementation gene M (FANCM), nonsense mutation c.5101C>T (p.Q1701X) was considerably extra frequent amongst breast most cancers sufferers than amongst controls [odds ratio (OR) = 1.86, 95% CI = 1.26-2.75; P = 0.0018], with explicit enrichment amongst sufferers with triple-negative breast most cancers (TNBC; OR = 3.56, 95% CI = 1.81-6.98, P = 0.0002).
In the Helsinki and Tampere areas, respectively, service frequencies of FANCM p.Q1701X had been 2.9% and 4.0% of breast most cancers sufferers, 5.6% and 6.6% of TNBC sufferers, 2.2% of ovarian most cancers sufferers (from Helsinki), and 1.4% and a couple of.5% of controls.
These findings establish FANCM as a breast most cancers susceptibility gene, mutations during which confer a notably robust predisposition for TNBC.